My research focuses on understanding the evolutionary dynamics and implications of admixture and introgression, that is, the mixing of populations that have been separated for relatively short and long evolutionary time, respectively. Given their central role in human evolution, I am interested in how they have shaped present-day genetic variation and disease risk, with a focus on structural variation. To do so, I am integrating population genetics and comparative genomics with bioinformatics tool development. My current research areas include:
Leveraging Methylation Information in Long-Read Sequencing Data To Improve Variant Phasing
Accurate phasing of genetic and epigenetic variation is crucial for many downstream analyses, including association testing, clinical variant interpretation, and population history inference. Although long-read sequencing significantly improves the continuity and completeness of genome sequencing, reconstructing chromosome-scale haplotypes still often requires combining multiple technologies, such as PacBio HiFi and Oxford Nanopore Technologies (ONT) sequencing. While these sequencing platforms detect the epigenetic modification 5-methylcytosine (5mC), current read-based phasing and de novo genome assembly algorithms do not incorporate this information. To assess whether methylation information can improve long-range phasing, I developed LongHap, a read-based phasing method that seamlessly integrates sequence and methylation data. LongHap outperforms existing tools by achieving lower error rates and greater phase block contiguity (Figure 1).
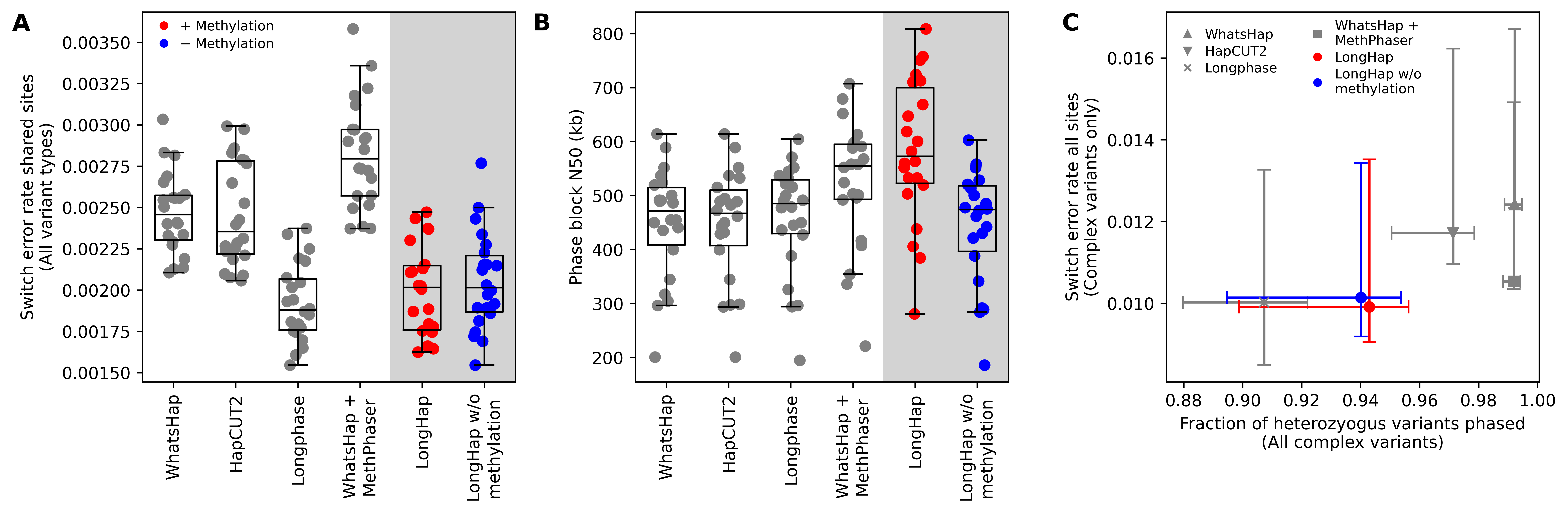
To extend these performance gains to phased de novo genome assemblies, I am now working to incorporate LongHap’s methylation-aware phasing module into an assembly polishing pipeline, improving haplotype accuracy in partially phased de novo genome assemblies generated from either PacBio HiFi or ONT sequencing data and reducing the need for multiple costly sequencing experiments that require large amounts of DNA.
Relevant work:
- Aaron Pfennig and Joshua M. Akey, Methylation-aware long-read phasing significantly improves genome-wide haplotype reconstruction, bioRxiv, https://doi.org/10.64898/2026.03.11.710820
Developing a tool suite for population and statistical genetics analyses on pangenomes
Pangenomes are a set of multiple whole-genome sequences typically represented as a graph (Figure 2). Unlike a single reference genome, pangenomes capture the full spectrum of genetic variation by being able to represent multiple mutations at the same position and accommodate substantial amounts of non-reference sequence.
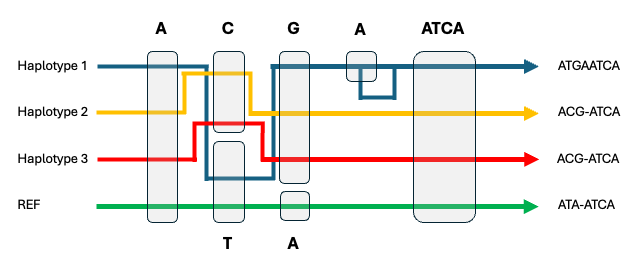
These capabilities are particularly critical for studying biomedically important highly diverse and structurally complex “genomic black holes” that are now accurately reconstructed in de novo genome assemblies.
However, due to an absence of analytical tools to analyze pangenomes, current approaches map genetic variation in pangenomes back to the coordinate system of a single reference genome and analyze it with conventional methods, which largely defeats the advantages of de novo assembly. Thus, the absence of analytical tools for pangenomes prevents us from taking advantage of the full spectrum of genetic variation (in particular, structurally complex variation) in population and statistical genetics analyses.
To address this, I am currently developing a tool suite to support standard population and statistical genetics analyses (e.g., elucidating population structure, association testing, etc.) on pangenomes.
New population genetics frameworks for analyzing structural variation
A major gap in human genetics is our inability to study structural variants (SVs), such as deletions and duplications ≥50 base pairs, that can disrupt protein-coding genes and regulatory elements, as they are invisible to short-read sequencing. This gap limits our understanding of disease and genome evolution. Long-read sequencing now enables the discovery of SVs through de novo assembly. However, SVs arise through complex mutational processes that defy basic population genetics assumptions, such as constant mutation rates, and we therefore lack foundational models to analyze this important variant class.
I am developing theoretical and analytical frameworks to make direct inferences about SVs by modeling their unique mutational mechanisms. For example, copy number variants can be modeled as chromosomes switching between “states” by gaining or losing copies. Two chromosomes can only share a common ancestor when they are in the same state (copy number). Such models inform realistic simulations to train machine learning models for making evolutionary and clinical inferences about SVs. By developing novel methods for assembling high-quality phased genomes and analyzing SVs, my research will delineate the functional impact of SVs.
Relevant work:
- Kwondo Kim, Aaron Pfennig (author 2 out of 12), …, and Charles Lee, A Denisovan-derived Alu insertion in OCA2 contributes to pigmentation diversity in present-day Melanesian, 2026, bioRxiv, https://doi.org/10.64898/2026.03.18.712481
Theoretical and empirical population genetics of admixture and introgression
Admixture and introgression play a central role in human evolution. Therefore, it is critical to understand how these processes have shaped the distribution of present-day genetic variation and what its implications are. I introduced a novel population genetics model that accounts for a heterogeneous genetic background in hybrid genomes, showing that fitness effects arising from a heterogeneous background modify the dynamics of focal alleles during introgression. The model predicts that most introgressed alleles must survive an initial filter, suggesting Neanderthal-introgressed variants in extant human genomes cannot be that harmful.
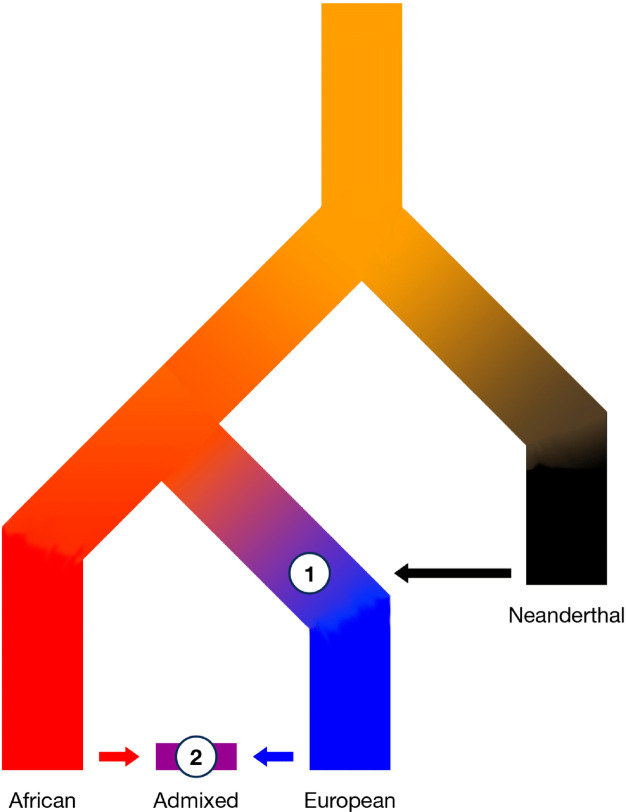
I empirically tested this hypothesis using 30,780 predominantly African- and European-like admixed genomes from the United States. In such genomes, the fitness of Neanderthal introgressed variants was re-evaluated as most Neanderthal introgressed sequence was contributed by European-like ancestors and brought onto a “novel” African-like genetic background (Figure 3). In concordance with predictions of my theoretical model, I found that remaining Neanderthal ancestry in contemporary genomes is likely evolutionary neutral as the admixed genomes contained approximately as much Neanderthal introgressed sequence as expected, suggesting widespread recent polygenic selection did not occur.
Furthermore, in related work, I proposed methods for including uncertainty in the interpretation of sex-biased admixture in the Americas in the context of the transatlantic slave trade. I collaborated on studies elucidating the genetic basis of prostate cancer and male pattern baldness in African men.
Relevant work:
†These authors are co-corresponding authors
- Kwondo Kim, Aaron Pfennig (author 2 out of 12), …, and Charles Lee, A Denisovan-derived Alu insertion in OCA2 contributes to pigmentation diversity in present-day Melanesian, 2026, bioRxiv, https://doi.org/10.64898/2026.03.18.712481
- Aaron Pfennig† and Joesph Lachance†, The evolutionary fate of Neanderthal DNA in 30,780 admixed genomes with recent African-like ancestry, bioRxiv, 2024, https://doi.org/10.1101/2024.07.25.605203 (under review at MBE)
- Aaron Pfennig and Joseph Lachance, Hybrid fitness effects modify fixation probabilities of introgressed alleles, G3 Genes|Genomes|Genetics, 2022, https://doi.org/10.1093/g3journal/jkac113
- Aaron Pfennig and Joseph Lachance, Challenges of accurately estimating sex-biased admixture from X chromosomal and autosomal ancestry proportions, The American Journal of Human Genetics, 2023, https://doi.org/10.1016/j.ajhg.2022.12.012
- Aaron Pfennig, Lindsay N Petersen, Paidamoyo Kachambwa, Joseph Lachance, Evolutionary genetics and admixture in African populations, Genome Biology and Evolution, 2023, https://doi.org/10.1093/gbe/evad054
- Rohini Janivara, Ujani Hazra, Aaron Pfennig (author 3 out of 22), …, and Joseph Lachance, Uncovering the genetic architecture and evolutionary roots of androgenetic alopecia in African men, Human Genetics and Genomics Advances, 2025, https://doi.org/10.1016/j.xhgg.2025.100428
- Burcu F. Darst, Raymond Hughley, Aaron Pfennig (author 3 out of 101), …, and Christopher A. Haiman, A Rare Germline HOXB13 Variant Contributes to Risk of Prostate Cancer in Men of African Ancestry, Eur Urol, 2022, https://doi.org/10.1016/j.eururo.2021.12.023